Data process
The processing of EuphausiiDB data is done by following the assembly and annotation workflows whose scripts are present in the euphausiiDB-bioanalysis directory.
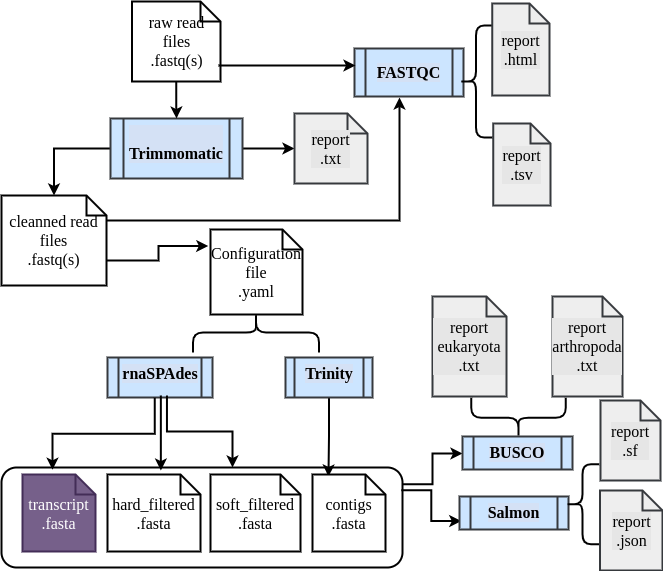
The transcriptome assembly workflow:
It includes 5 distinct steps:- Quality evaluation of raw data with FastQC;
- Raw data processing with Trimmomatic to filter and trim reads according to their sequence quality;
- Quality evaluation of cleanned data with FastQC;
- De novo assembly step using rnaSPAdes / Trinity ;
- Quality evaluation of the assembled transcripts using BUSCO and salmon .
Github repository:
- Assembly pipeline for paired-end reads: Transcriptome Assembly
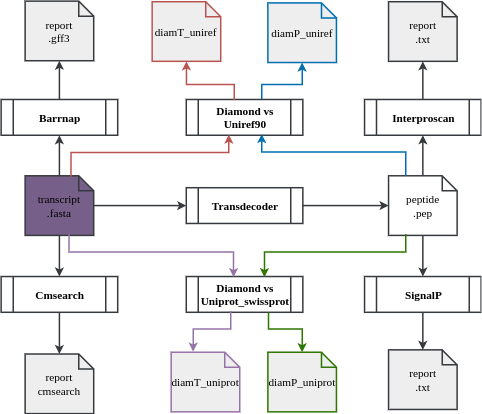
The annotation workflow:
Downstream analyses of assemblies includes:- Transcriptome completion evaluation using Busco;
- Prediction of ribosomal RNA gene locations in transcripts
- Diamond a sequence aligner for protein and translated DNA searches against Uniref90 and uniprot-swissprot databases
- Prediction of coding regions prediction using TransDecoder;
- cmsearch uses the covariance model (CM) in cmfile to search for homologous RNAs in seqfile, and outputs high-scoring alignments
- Functional annotation of predicted proteins using the InterProscan pipeline from EMBL-EBI.
PS: as of assembly 018, there are no associated KEGG terms because InterProscan version 5.59-91.0 does not include them in the annotation
Github repository:
- Annotation pipeline for paired-end reads: Transcripts Annotation